
18.5 Understanding fungal genetic structure
We have a long heritage of using fungi and fungal products. Some of our current fungal biotechnology, such as baking, brewing and the numerous fermented food products described in Chapter 17, originated hundreds or even thousands of years ago and largely by the chance association between natural fungi and one or more of the constituents of the food material. Although the original discovery of penicillin was also a matter of chance (Section 17.15) its industrial production in the middle of the 20th century was a much more directed process, as was the development of other products such as citric acid (Section 17.14). Yet all improvements were at the organismal level. Techniques were found that enabled cultivation of specific organisms, and strains were selected that had advantageous biological characteristics.
In the second half of the 20th century the rapidly accumulating knowledge of fungal genetics was brought to bear, and from what we have discussed so far it must be clear that a thorough understanding of the molecular genetics of fungi is essential to future exploitation. The basic genetic architecture of fungi is typical of eukaryotes in general (Section 5.6). All the major principles of eukaryote genetics apply in fungi; gene structure and organisation, Mendelian segregations, recombination, and the rest of the meiosis-dependent features. These are aspects of what might be called Mendelian or segregational genetics, which apply because of the chromosomal architecture and mechanisms of meiosis (Moore & Novak Frazer, 2002; Dyer et al., 2017).
Chromosome maps constructed solely from recombination frequencies have a limited resolution, although large numbers of progeny can be scored in most microorganisms, so reducing this problem to some extent. When the Saccharomyces cerevisiae genome sequencing project began in 1989, the conventional genetic map consisted of more than 1,400 markers, an average of one every 3.3 kb, and this was detailed enough for the sequencing programme without the need for much more physical mapping. However, Saccharomyces cerevisiae was one of the two most intensively mapped eukaryotes at the time (the fruit fly, Drosophila, being the other), so physical mapping is necessary to improve the marker density in other fungi as they are included in genome sequencing programmes.
Physical mapping procedures include restriction mapping, which establishes the positions of restriction endonuclease recognition sites in a DNA molecule; locating markers on chromosomes by hybridising marker probes to intact chromosomes; and mapping known sequences in genome fragments using PCR and hybridisation. The ideal is to establish the locations of unique sequences, which are not duplicated at any other site, as markers spaced about 100 kb apart (that’s just less than 1% genetic recombination) throughout the genome. The collection of such markers is known as a mapping panel.
Use of mapping panels is essentially a management technique for sharing the effort between participating laboratories, something which has been common for many years in several physical sciences, like particle physics and astronomy, though genome sequencing represented the entry of biology into the ‘big-science’ league. The first example of big fungal science was the programme to sequence the yeast genome which was initiated in 1989 by the European Commission. The project involved 35 European laboratories at the outset, and the first sequence of a complete chromosome was published in 1992. Eventually, over 600 scientists were involved, at locations in Europe, North America and Japan, and progress involved distribution of DNA fragments to the contributing laboratories by the DNA coordinator. The complete sequence of the yeast genome was published in 1997.
The genome is made up of the entire DNA content of a cell. Eukaryotes and prokaryotes have quite different types of genome, but it is generally assumed that something like the prokaryotic grade of organisation is the primitive form from which the eukaryote organisation evolved. Modern prokaryotes and eukaryotes have a great deal in common (see Chapter 5); the DNA of a gene is transcribed into RNA, which is called a messenger RNA (mRNA) if it is a transcript of a protein-coding gene, and the mRNA is translated into protein by the ribosomes and other translation machinery. The part of a protein-coding gene sequence that is translated into protein is called the open reading frame, usually abbreviated to ORF.
As a genome sequence is assembled the functional genes in the sequence are recognised as open reading frames (ORFs); the process is called genome annotation and is discussed in more detail below. Not all the ORFs that are identified can be associated with a gene of identified function; an ORF specifying a product that does not resemble a known protein is called an unidentified reading frame, or URF. But comparative genomics does more than identify the genes. It can show the evolutionary relationships between different organisms, and aids understanding of how the genotype relates to life-style and environment.
Characteristically, the ORF is read in the 5' to 3' direction along the mRNA, and it starts with an initiation codon and ends with a termination codon (Fig. 14). Nucleotide sequences that occur in the mRNA before the ORF make up the leader sequence, and sequences following the ORF make up the trailer segment. Many eukaryotic genes are split into exons (meaningful segments) and introns (sequence segments that do not contribute to the protein-coding sequence).
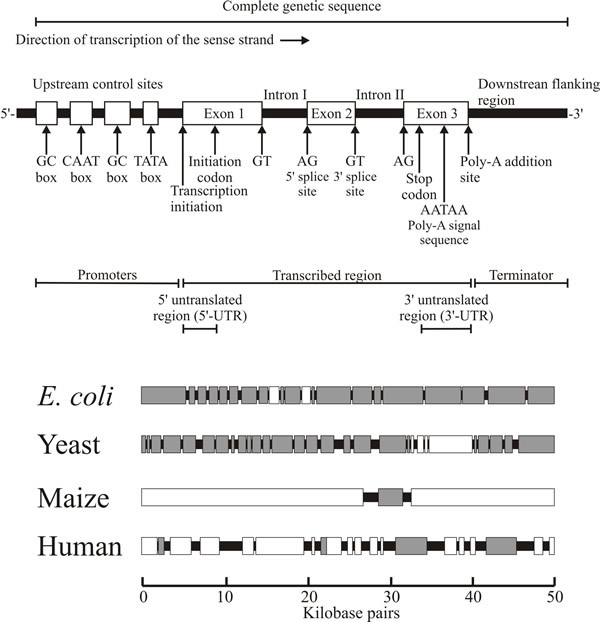 |
| Fig. 14. Top: the basic structure of a typical eukaryotic gene. The schematic diagram indicates the structure of a type II gene; that is, a protein-encoding gene transcribed by polymerase II. The diagram is not drawn to scale and the relative sizes of the different sections differ between genes and between the eukaryotic Kingdoms. Bottom: comparison of 50 kbp segments of the genomes of the prokaryote Escherichia coli and three eukaryotes to show how the ‘density’ of genetic information varies. In each case the grey boxes correspond to gene sequences, and the white boxes correspond to stretches of repeated sequences. Adapted from Moore & Novak Frazer, 2002. |
The introns are removed from the primary RNA transcript by the splicing machinery to form the functional mRNA (Fig. 14; and see Section 5.4), and several different mRNAs may be produced from any one primary RNA transcript by a process known as alternative splicing. And then there are the transposable elements, often called transposons, which are seemingly ubiquitous genetic elements that have been discovered in all prokaryotes and eukaryotes so far investigated. In remarkable contrast to all other genes, transposable elements are able to move to new locations within their host genomes in a process called transposition. Transposable elements contribute enormously to eukaryotic genome diversity. Their ubiquitous presence affects the genomes of all species; mediating genome evolution by causing mutations, repetitions and chromosomal rearrangements and by modifying gene expression.
Indeed, introns and transposons seem to be extremely ancient genetic structures, which certainly existed long before the eukaryote grade of organisation emerged. So, over evolutionary time they have created new patterns of gene expression by ‘shuffling’ functional motifs together and then combining them with new control elements to produce differentiated cellular structures with new morphologies and/or new developmental possibilities. And not just within the one genome. Transposition is also responsible for the horizontal gene transfer events of the sort we describe in Section 17.15 in relation to penicillin biosynthesis. Remember that horizontal gene transfer is the transmission of genetic material between organisms and across major taxonomic boundaries and that there is evidence for many horizontal gene transfer events in fungi (Richards et al., 2011; Slot et al., 2017; Steenkamp et al., 2018). All of this is discussed further below.
The smallest eukaryotic genomes, of some yeasts, are in the region of 10 Mb (Mb is the usual abbreviation for a million base pairs or a 'megabase'), and the largest (in vertebrates and plants) are over 100,000 Mb, so we can observe some surprising structural differences when we compare different eukaryotes (see Table 5.2 in Section 5.7). Generally speaking (but remember there are exceptions to all generalisations), it appears that space is saved in the genomes of less complex organisms by having the genes more closely packed together and by having much less repetition (Fig. 14, above).
The genome of Saccharomyces cerevisiae contains more genes per unit length of DNA than occur in human or maize DNA. On the other hand, up to 40% of the gene sequences of Saccharomyces cerevisiae are duplicated. In most cases the duplicated sequences are so similar that their protein products are identical and, presumably, either functionally redundant, or (more likely) under very different regulatory control. Furthermore, as we detail in Section 18.10, below, the whole of Saccharomyces cerevisiae chromosome XIV is made up of regions duplicated on other chromosomes. Nevertheless, the small size of the yeast genome is one reason why yeast geneticists and molecular biologists pioneered eukaryote genome analysis.
Although some of the more unusual aspects of genome structure observed in higher animals and plants might not be represented in fungi, the genomes of yeast and other fungi remain good models of eukaryotic genetic architecture and their smaller size means that the information they contain is technically more accessible. In terms of genetic information content, the organisation of the fungal genome is, generally, much more economical than that of animals and plants. Although fungal genes may be generally more compact with fewer introns and shorter spaces between genes, a major difference is that fungi contain much less of the so-called redundant DNA, which is devoted to repetitive noncoding sequences in so many animals and plants. But then, fungi have made specialist use of heterokaryosis (see Chapter 7), which is uncommon in the other kingdoms. What better way is there to increase genome size than being a syncytium?
So, despite the differences that undoubtedly exist, fungi are typical eukaryotes, featuring all the basic cell biology expected of this grade of organisation. Even though the yeast genome is only in the same size range as some of the more advanced prokaryotes, the genetic structure and functioning of genes of filamentous fungi are representative of all eukaryotes and we can use their sequences to learn about genomics (Moore & Novak Frazer, 2002).
Analysis of the genetic sequences that make up the genome of an organism, and comparisons of the genomes of different organisms (exercises that have come to be known as the science of genomics) only became possible from the mid-1990s. Establishing the exact DNA sequence of a genome is a major undertaking, but is only the prelude to intensive analysis. The priority of genomics is to establish the number and function of genes in an organism.
In Section 18.10 we will describe the methods developed for sequencing and studying whole fungal genomes, but first we want to discuss a few more fungus-specific genetic details; namely introns, alternative splicing, transposons, genomic variation, including gene clusters and horizontal transfer, and ploidy variation (and see Sánchez et al., 2020).
Updated January, 2020