
18.1 Antifungal agents that target the membrane
Because fungi are themselves eukaryotes, drug discovery programmes aimed at finding antifungal agents with the required specificity must concentrate on targeting features that are unique to fungi. Attention must also be given to the type of material being researched. By convention fungicides are used to treat plants, whilst antifungal drugs are used to treat animals. Either of these may be (a) fungistatic, meaning that the agent stops fungal growth without killing the fungus and thereby ‘buys time’ for the diseased organism’s intrinsic defences to operate; or fungicidal, meaning that the agent is fatally toxic to the fungus, without, hopefully, being in the least damaging to the diseased organism.
A protectant fungicide or antifungal drug is a chemical that acts outside the plant or animal and protects it from fungal infection. Protectants usually have multiple sites of action:
- Milardet's ‘Bordeaux mixture’ (a paint-like mixture of calcium hydroxide and copper sulfate) was originally developed as a fungicide in vineyards to control downy mildew on grape vines, is also used to protect potato plants from Phytophthora infestans infection and is an effective protectant of all sorts of plants against all sorts of stem and foliar fungal and fungus-like diseases.
- Whitfield's ointment (a greasy mixture of benzoic and salicylic acids) is used to treat athlete's foot and is effective against similar superficial disease fungi.
A compound like Bordeaux mixture affects a range of sensitive sites in the fungus but its mode of action at some of these sites may be the same (it is the copper ion which is fungitoxic). The contrast here is between an antifungal that affects a single fungal site, which hopefully is unique, and an antifungal that affects several sites in the fungus. The former will exhibit more selective toxicity than the latter. On the other hand, resistance of the fungus to the latter type of antifungal will develop much more slowly than to the former. So, it’s a trade-off between reduction in crop yield, because of lower selective toxicity (for which aspect single site antifungals perform better than multiple site ones), and the rate at which resistant strains arise in the fungal population (for which aspect multiple site antifungals perform better than single site ones). Of course, selective toxicity is more important for antifungal drugs than for agricultural fungicides.
Systemic fungicides and antifungal drugs penetrate the plant or animal tissues from the site of application and spread through the tissues of the host, eliminating established infections. Systemic antifungal agents usually have a highly specific (generally single) site of activity, for example:
- benzimidazole, targets microtubules of fungal pathogens of plants,
- ketoconazole, targets sterol biosynthesis of fungal pathogens of animals,
- and see Table 1.
Table 1. Overview of the sites
of action of some systemic antifungal agents |
|||
|---|---|---|---|
Cellular target |
Host (i.e. diseased) organism |
Agent |
Mode of action |
| Plasma membranes | animals |
Polyenes (Fig. 2) |
bind to ergosterol |
animals & plants |
Azoles (Figs 5 & 6) |
inhibit ergosterol synthesis |
|
plants |
Edifenphos (an organophosphorus fungicide) (Fig. 11) |
inhibits phosphatidyl choline synthesis |
|
| Nucleic acid biosynthesis | plants |
Acylalanines (e.g. metalaxyl) (Fig. 11) |
inhibit RNA polymerase |
animals |
5-fluorocytosine |
inhibits RNA synthesis |
|
Mitochondrial respiration |
plants |
Strobilurins (Figs 12 & 13) |
bind to cytochrome b, disrupting electron flow and ATP synthesis |
| Cell wall | plants |
Polyoxins, Nikkomycins (Fig. 8) |
inhibit chitin synthesis |
plants |
Tricyclazole (Fig. 11) |
inhibits melanin synthesis |
|
man |
Echinocandins (Fig. 9) |
inhibit β 1-3 glucan synthase |
|
| Cytoskeleton | plants |
Benzimidazoles (Fig. 10) |
bind to β-tubulin |
animals |
Griseofulvin (Fig. 10) |
binds to microtubule associated proteins |
|
| Other potential targets include: mannoproteins involved in adhesion, translation elongation factor, and proteases. | |||
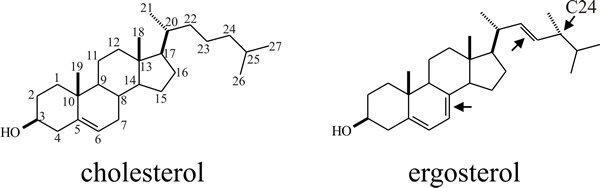
The basis of the selective toxicity of antifungal agents that target plasma membranes is the relative sterol composition of those membranes (see the Plasma membrane and signalling pathways section in Chapter 5 and The fungal wall as a clinical target in Chapter 6; CLICK on the section titles to view the pages now). Sterols influence the fluidity, permeability, microdomain formation, and membrane activities including functionality of membrane-bound proteins. The key fact relevant to antifungal drug discovery is that there are three predominant forms of sterol found in eukaryotes: cholesterol in animals, phytosterols (sitosterol, stigmasterol, campesterol) in plants, and ergosterol in fungi. The ergosterol biosynthesis pathway is fungus-specific (Fig. 1, below) so it is the prime target for treating fungal diseases in animals and plants alike.
Resources Box Information about toxicity Rapid access to internationally peer reviewed information on chemicals commonly used throughout the world, including those that occur as contaminants in the environment and in food, is provided by the INCHEM website of the International Programme on Chemical Safety. It consolidates chemical safety information from a number of intergovernmental organisations whose goal it is to assist in the sound management of chemicals. View this URL: http://www.inchem.org/ |
 |
|---|
| Fig. 1. Chemical structures of cholesterol and ergosterol. The cholesterol schematic shows the conventional numbering scheme for the carbon atoms. Ergosterol differs from cholesterol by having an extra methyl group at C24 and two additional sites of unsaturation (arrows). |
Some fungi do not contain ergosterol (some exceptions are noted in Section 5.14), including chytrids (which use cholesterol) and the basidiomycete Puccinia graminis (which uses fungisterol). The fungus-like Leptomitales and Saprolegniales (Phylum Oomycota, Kingdom Straminipila; see Section 3.10) use cholesterol and other sterols like demosterol. Phytophthora cactorum is also an interesting exception in that it does not synthesise sterols and must obtain them from its substrate.
The two main classes of antifungals currently available for treating systemic mycoses target ergosterol. Polyenes act by selective disruption of membrane structure by binding to ergosterol, causing membrane permeability. These drugs have toxicity and pharmacological difficulties. The azoles inhibit the lanosterol demethylase step in ergosterol biosynthesis and thereby hinder membrane synthesis in fungi, but use of these agents is being affected by the emergence of azole-resistance.
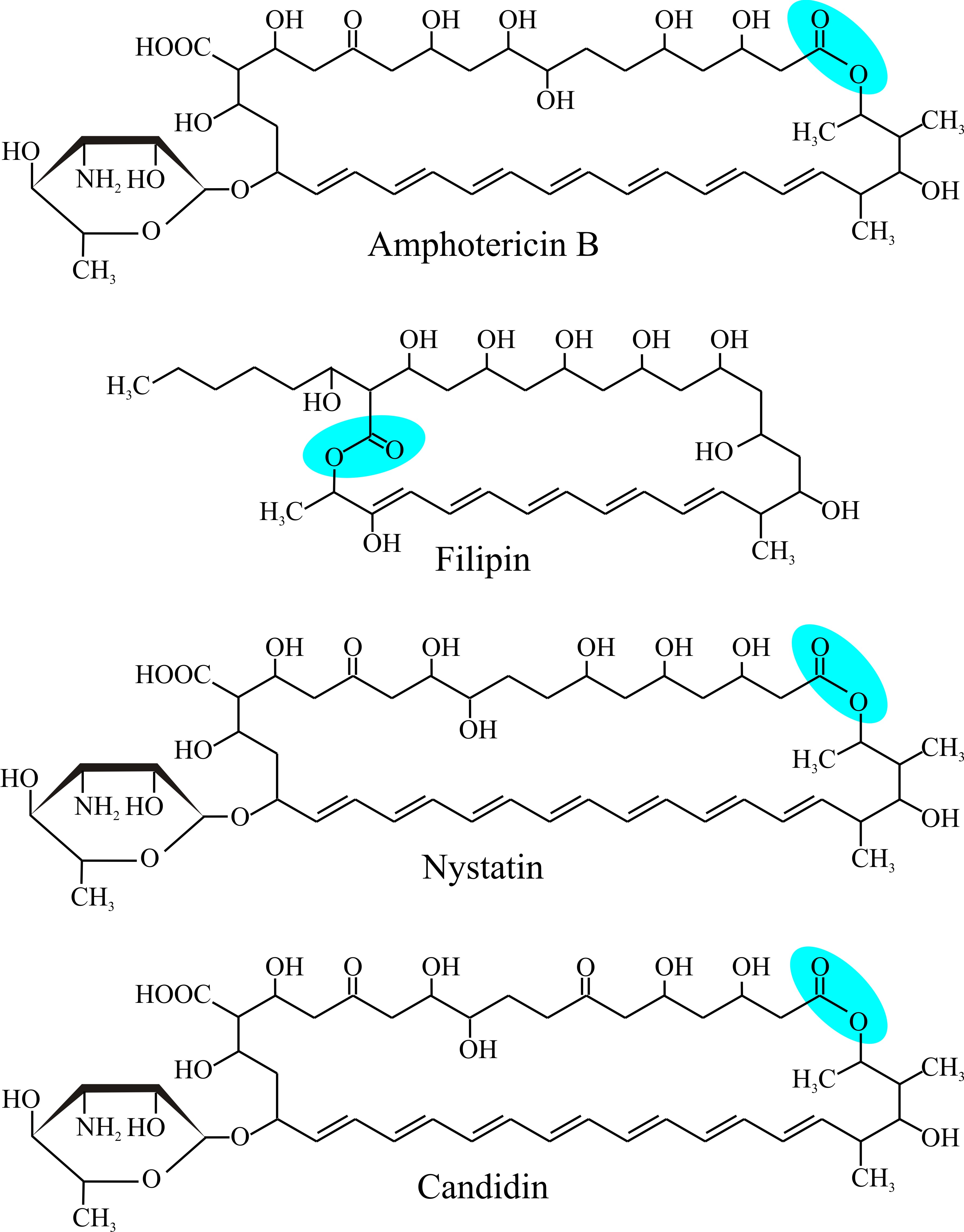
Polyenes are compounds that contain alternating double and single carbon-carbon bonds. Compounds with two carbon-carbon double bonds alternating with single bonds (−C=C−C=C−) are conjugated compounds called dienes; those with three such double bonds are trienes (−C=C−C=C−C=C−); those with four are tetraenes, etc. Compounds with one or more sequences of many alternating double bonds are the polyenes (Fig. 2).
 |
|---|
| Fig. 2. Structural formulae of some polyene antibiotics. The top illustration shows Amphotericin B, and should be compared with Fig. 3 as an example of a clinically-important polyene antifungal agent. Note the alternating double and single bonds (conjugated double bonds all trans) across the bottom of all the molecules as oriented here and the lactone closure (−(C=O)−O−) of the macrolide ring (blue panel), and the mycosamine amino sugar at bottom left. Filipin lacks the hexosamine carbohydrate. |
Polyenes are terpenes which are derived biosynthetically from units of isoprene (C5H8) (see Fig 15 in Chapter 10). These are polyunsaturated organic compounds and the category includes many fatty acids (hence the description ‘polyunsaturated fat’). Normally, carbon-carbon double bonds are sufficiently energetic to absorb light in the blue and ultraviolet region of the spectrum, resulting in compounds which are coloured yellow or orange. Consequently, many pigments feature linear polyenes, for example the yellow-orange coloured β-carotene (see Fig 20 in Chapter 10); and polyene antifungals tend to be coloured yellow.
Nystatin (Fig. 2) was isolated from the bacterium Streptomyces noursei in 1950 as a promising antifungal agent, and amphotericin B was isolated from Streptomyces nodosus a few years later; in fact, of more than 200 polyenes antibiotics so far discovered, the majority are produced by streptomycetes (Table 2). Nystatin is used topically (that is, applied to body surfaces; for example, to control vulvovaginal candidiasis) or orally (for example to control alimentary tract infections) as there is little or no absorption by the alimentary tract, but it is too toxic to be used by intravenous or intramuscular injection (that is, it is too toxic to be administered parenterally). Amphotericin B is the only polyene which is sufficiently non-toxic to be tolerated parenterally although side effects from prolonged exposure include kidney damage and nausea.
Table 2. Original sources of
some polyenes that have been used in experimental and/or clinical
settings |
|
|---|---|
Name |
Producing organism |
Amphotericin B (isolated in 1955) |
Streptomyces nodosus |
Candicidin |
S. griseus |
Candidin |
S. viridoflavus |
Etruscomycin (lucensomycin) |
S. lucensis |
Filipin complex (isolated in 1955) |
S. filipensis |
Hamycin |
S. pimprina |
Natamycin (pimaricin) |
S. natalensis |
Nystatin (isolated in 1950) |
S. albidus or S. noursei |
Trichomycin |
S. hachijoensis and S. abikoensis |
There’s more to antifungal polyenes than just the conjugated polyene structure. They are characterised by a macrolide ring of carbon atoms (a macrolide is a large macrocyclic structure) which is closed into a ring by formation of a lactone (a lactone is a cyclic ester resulting from condensation of an alcohol group and a carboxylic acid group in the same molecule) and to which a 6-carbon deoxyaminosugar is characteristically attached. With one exception, the hexosamine carbohydrate is the deoxyaminosugar mycosamine (Fig. 2).
As you can see from Fig. 2, amphotericin B has seven hydroxyls and the mycosamine sugar on one side of the macrolide ring (which is hydrophilic as a result) and the double-bonded polyene on the other side of ring (and this side is hydrophobic as a result); and as its name implies, the molecule is amphoteric (capable of reacting chemically either as an acid or as a base). These structural features are thought to determine the sterol selectivity and biological activity of the molecule. Polyenes have limited solubility in water but readily interact with the sterol molecules or acyl side chains of phospholipids in biological membranes. This is the basis of their biological activity because a result of their interaction with membrane sterols is the formation of polyene-sterol complexes that can produce pores (partial or complete) through the cell membrane so that membrane permeability is increased.
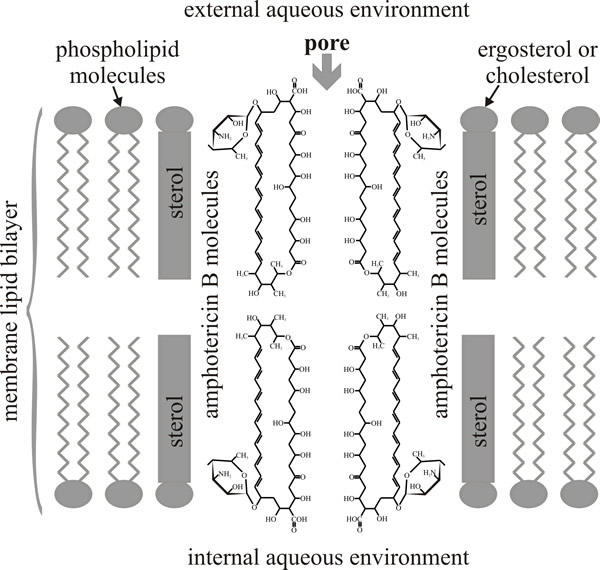
Amphotericin B forms an annulus, which has a pore of 0.8 nm internal diameter, of 8 polyene molecules in one leaflet of the membrane bilayer. This is a ‘half-pore’ but the annuli can form in both leaflets and when two annuli coincide in both opposing leaflets the amphotericin pore spans the lipid bilayer as an unregulated ion channel. The orientation of the aminosugar on the macrolide ring determines sterol preference, and specific hydrogen bonding (between 2'-OH of amphotericin and 3β-OH of ergosterol) locks the complex in place so that eight of them enclose the hydrophilic channel (Fig. 3).
|
|---|
| Fig. 3. Diagrammatic structure of an amphotericin B-sterol pore. The illustration depicts the alignment of two half-pores; the half-pores are assembled from eight molecules of amphotericin in a ring in each leaflet of the lipid bilayer. When two half pores coincide in both leaflets of the membrane bilayer the hydrophilic channel in the centre forms an unregulated ion channel that spans the membrane. |
Release of cellular K+ is one of the earliest effects of polyenes and this is associated with uptake of protons and consequent acidification of cytoplasm. Leakage of amino acids, sugars and other metabolites also occurs (Table 3).
Table 3. Observable effects of
four polyenes that interact with membrane sterols |
||||
|---|---|---|---|---|
Feature |
Filipin |
nystatin |
amphotericin B |
Candicidin |
Potassium leakage |
+ |
+ |
+ |
+ |
K+ or NH4+ reversal* of glycolysis inhibition |
- |
- |
+ |
+ |
Phosphate leakage |
+ |
+ |
± |
± |
Erythrocyte lysis |
Rapid |
+ |
+ |
± |
Yeast protoplast lysis |
Rapid |
+ |
± |
- |
Antifungal activity |
+ |
++ |
+++ |
++++ |
| *The ammonium ion is physically sufficiently similar to the potassium ion to serve as a potassium analogue in many chemical reactions. There are many similarities between potassium and ammonium salts. | ||||
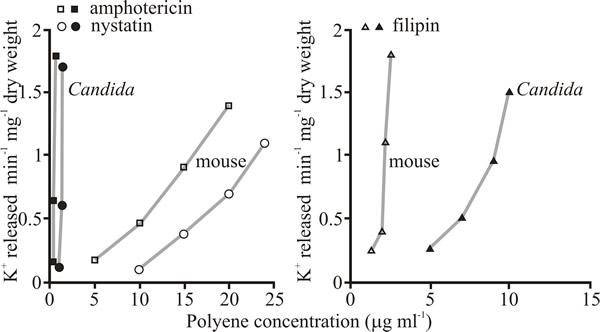
Polyenes interact with both cholesterol and ergosterol so the key to their value as clinically useful antifungal agents is the fact that some have so much more affinity for ergosterol that they can be considered to be selectively toxic to the fungus (Fig. 4 and Table 4). Others, like filipin, are too toxic to the host animal to be clinically useful (and see Table 4). Filipin does not form ion channels like amphotericin B and nystatin but acts as a membrane disrupter. Although it is antifungal, it is more toxic to animal cells than fungi (Fig 4) because of its affinity for cholesterol. However, this affinity for cholesterol and the fluorescence of the polyene have made filipin an extremely useful histochemical stain for cholesterol.
 |
|---|
| Fig. 4. Selective toxicity of polyene antifungals against Candida albicans illustrated as their effect on release of potassium ions from intact Candida cells compared with release from mouse fibroblasts. Left hand panel compares the methyl ester of amphotericin B and nystatin with mouse fibroblasts (both of these polyenes have higher affinity for ergosterol than for cholesterol) and the right hand panel shows the relative effect of the polyene filipin, which lacks an aminosugar in its chemical structure (Fig. 2) and has much greater affinity for cholesterol than it does for ergosterol. |
Table 4. Toxicity of polyenes
in small mammals* |
|||||
|---|---|---|---|---|---|
| Antibiotic | LD50 (mg kg-1) |
animal |
|||
intravenous |
intraperitoneal |
subcutaneous |
oral |
||
Amphotericin B |
4-6.6 |
280-1 640 |
|
>8 000 |
mouse |
Candicidin |
|
2.1-7 |
160-280 |
98-400 |
mouse |
Nystatin |
3 |
45 |
2.4 |
8 000 |
mouse |
Pimaricin (natamycin) |
5-10 |
250 |
5 000 |
1 500 |
rat |
| *Compare these values with the median minimum inhibitory concentrations (MIC) against Candida albicans of: Amphotericin B, 0.5 mg l-1; Candicidin, 0.5 mg l-1; Nystatin, 3 mg l-1; and Pimaricin (natamycin), 5 mg l-1. | |||||
Enclosing amphotericin B within liposomes enhances drug delivery while reducing toxicity. Liposomes are artificial lipid vesicles (less than 100 nm diameter) made from readily available phospholipids, such as hydrogenated soybean phosphatidylcholine or egg phosphatidylethanolamine. The phospholipids form into ‘bubbles’ of membrane bilayers that can be used to encapsulate drugs which are poorly-soluble in water. The liposome membranes merge with cell membranes and therefore greatly improve delivery of the insoluble drug. In rabbit and mouse trials, Amphotericin B liposomes (called AmBisome) were more effective against Candida and Aspergillus than conventional Amphotericin B preparations; and, compared to Amphotericin B, the animal-toxicity of AmBisome was reduced by two thirds (Table 5); AmBisome is used to treat serious, life-threatening fungal infections of humans.
Table 5. Activity of
conventional amphotericin B (AmB) and the AmBisome (liposome)
preparation |
||
|---|---|---|
| Fungus | Drug preparation |
MIC90 (mg l-1) |
Candida albicans |
AmB |
1.25 |
AmBisome |
0.62 |
|
Candida tropicalis |
AmB |
1.25 |
AmBisome |
0.62 |
|
Aspergillus sp. |
AmB |
2.50 |
AmBisome |
1.25 |
|
Fusarium sp. |
AmB |
2.50 |
AmBisome |
2.50 |
|
| MIC90 = minimum concentration needed to cause 90% inhibition. | ||
Resistance to polyenes is rarely observed clinically although it can be demonstrated in the laboratory. Combined therapy reduces the risk of drug resistant strains emerging during antifungal therapy. Amphotericin B can be used in combination with flucytosine (5-fluorocytosine, a fluorinated pyrimidine analogue that targets fungal RNA synthesis and disturbs translation of essential proteins) for a beneficial effect.
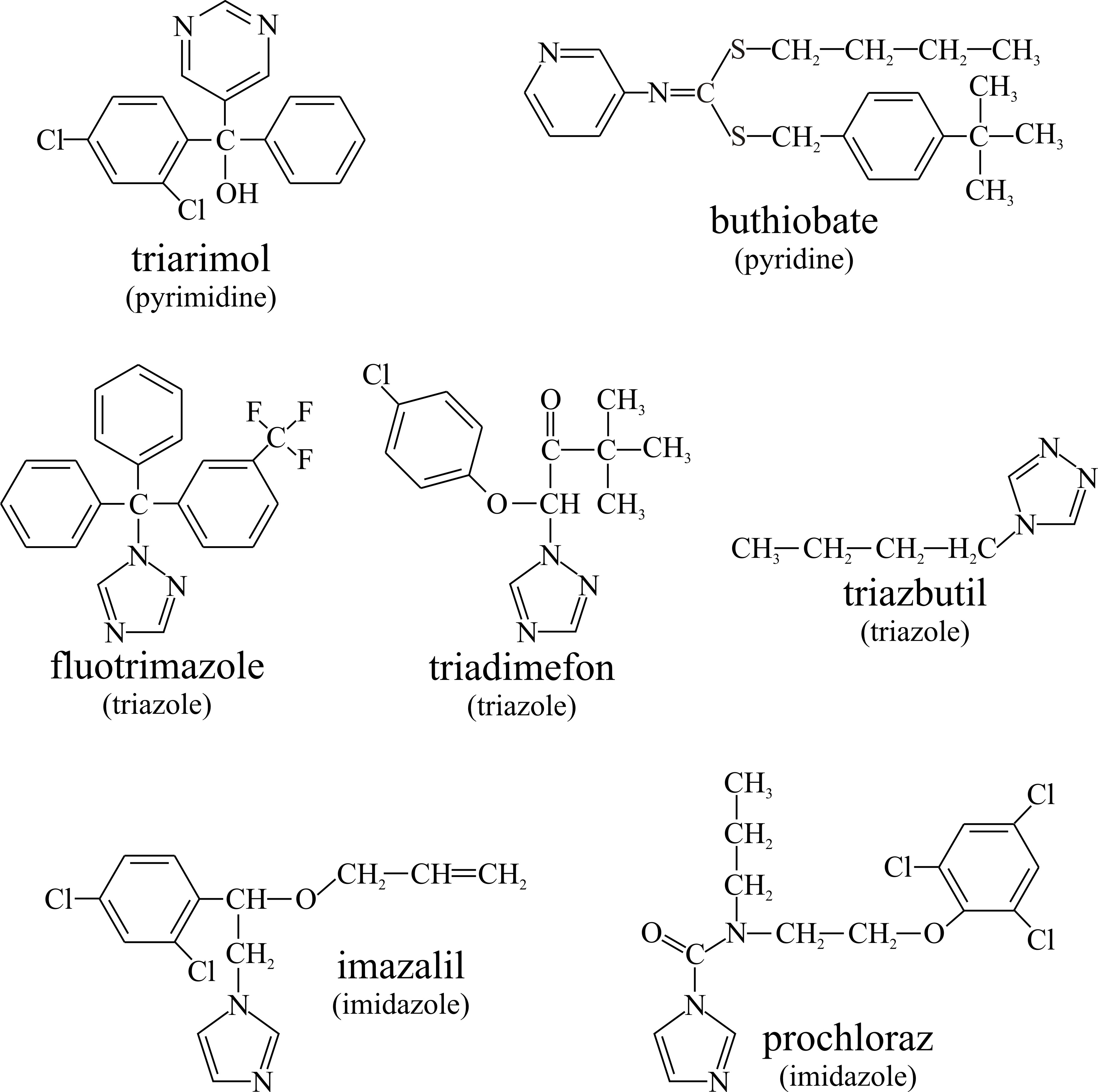
The polyenes interact with ergosterol to cause membrane leakage, but a range of other antifungal agents inhibit ergosterol biosynthesis and thereby reduce or even completely deplete the ergosterol content of the fungal membrane (Alcazar-Fuoli et al., 2008; Veen & Lang, 2005). Some of these compounds are used as fungicides to treat plant diseases (structural formulae shown in Fig. 5):
- Pyrimidine analogues (for example, triarimol),
- Pyridines, such as buthiobate,
- Triazoles (for example, fluotrimazole, triadimefon and triazbutil), and
- Imidazoles (for example, imazalil (= enilconazole) and prochloraz).
|
||
|---|---|---|
| Fig. 5. Some fungicides that inhibit ergosterol biosynthesis and thereby reduce or even completely deplete the ergosterol content of fungal pests. |
Similar compounds are used clinically as antifungal drugs (Fig. 6):
- Imidazoles like miconazole (relatively insoluble so used in ointments for topical application), clotrimazole and ketoconazole (relatively soluble and can be taken by mouth),
- Triazoles like itraconazole and fluconazole.
|
||
| Fig. 6. Structural formulae of some clinically useful antifungal azoles. |
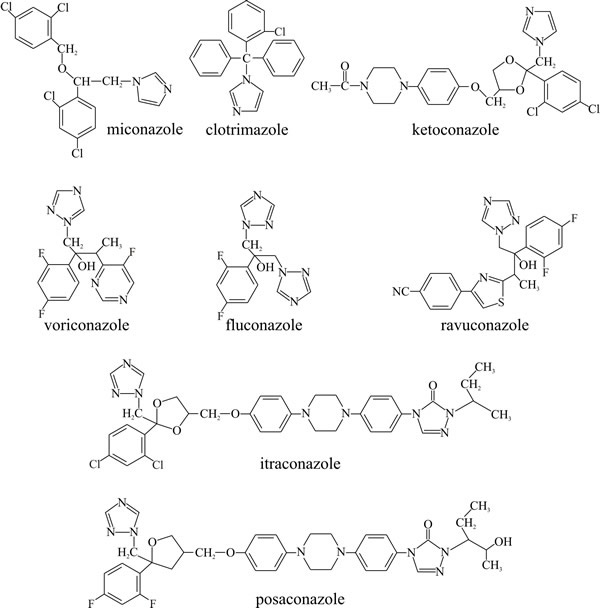
The characteristic feature of azoles is a five membered heterocyclic ring; in imidazoles this ring includes two nitrogen atoms. These were first discovered in the early 1970s and are used to treat topical (miconazole) and systemic (ketoconazole) infections of man. The triazole five membered ring contains three nitrogen atoms. Triazoles, which were first used in the 1990s, have lesser side effects compared to the imidazoles. Fluconazole and itrasconazole are highly soluble but are excreted renally more rapidly than ketoconazole and have a lower antifungal activity. These compounds affect ergosterol biosynthesis by binding to cytochrome P450. This enzyme is required for the demethylation of lanosterol. Its inhibition hampers membrane formation and leads to accumulation of steroid intermediates which destabilise the membrane and inhibit growth (the agents are fungistatic). Potency of imidazoles is determined by:
- the affinity and geometric orientation of the heterocyclic nitrogen ring to the heme iron atom in cytochrome P450
- protonation of the N-3 or N-4 in the imidazole or triazole ring
- affinity of the nonligand portion of the drug for the apoprotein of cytochrome P450.
An unprotonated nitrogen atom is needed to bind the haeme iron of cytochrome P450. The intracellular pH of the hyphal tip is usually less than 6 and at this pH the triazole ring of itraconazole is unprotonated whereas the imidazole ring of ketoconazole is more than 75% protonated. So itraconazole is a more potent antifungal drug than ketoconazole. Protracted use of ketoconazole can cause liver damage and fluconazole, which does not affect the liver, has more or less replaced ketoconazole for this reason. However, fluconazole has less antifungal activity than ketoconazole, so in AIDS patients with severe fungal infections and impaired immune systems the risk of ketoconazole liver damage can be worth taking.
When azoles bind to the cytochrome P450 demethylase, which is responsible for removal of methyl groups in biosynthesis of ergosterol, the 14-alpha-methyl (lanosterol) sterol content of the membrane increases, and the ergosterol content of the membrane decreases and properties of the membrane consequently deteriorate (Fig. 7). Ironically, one of the results of membrane deterioration is that chitin synthesis increases; but it is an uncoordinated increase and does not benefit the wall.
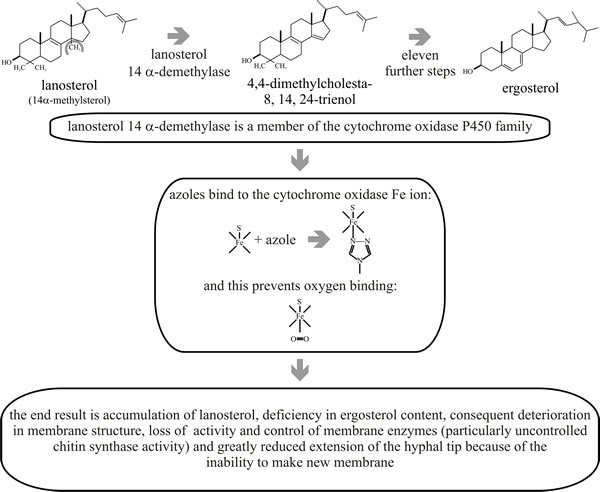 |
| Fig. 7. Mode of action of ergosterol biosynthesis inhibitors and effects on membrane function and cell proliferation. |
Table 6. Inhibition of
ergosterol synthesis by ketoconazole and itraconazole |
||
|---|---|---|
| Organism | Molar concentration causing 50%
inhibition of ergosterol synthesis |
|
Ketoconazole |
Itraconazole |
|
Candida albicans |
2.5 × 10-8 M |
5.0 × 10-9 M |
Pityrosporum ovale (a lipophilic yeast which is a normal component of human skin flora but can be an opportunistic pathogen) |
5.6 × 10-7 M |
5.0 × 10-9 M |
Aspergillus fumigatus |
5.6 × 10-7 M |
2.3 × 10-8 M |
Sterol biosynthesis is complex, with several branched and potential alternative pathways. The relative importance of each part of the pathway differs between species of fungi and this accounts for differences in their sensitivity to individual azoles (Table 6) (Cowen, 2008; Monk & Goffeau, 2008; Howard & Arendrup, 2011).
Resistance to fluconazole can arise due to:
- increased efflux of a drug from the cell resulting from mutations causing constitutive upregulation of multidrug transporter(s);
- alteration or amplification of the drug target, which minimises the impact of the drug on the cell.
- A third category of resistance mechanism involves cellular alterations that minimise toxicity of the drug, for example by upregulation of biosynthetic genes producing excess sensitive protein so that the inhibitory agent is effectively titrated out, or by secondary mutations that prevent accumulation of toxic intermediates (Da Silva Ferreira et al. 2005; Cowen, 2008). We will discuss below (Section 18.3) the potential of combinatorial therapy in lengthening the clinically useful lifetime of established drugs by reducing the evolution of resistance.
Voriconazole is the recommended agent for invasive aspergillosis in humans, with lipid amphotericin B or caspofungin as alternating treatments. As the only agents available in oral formulations, azoles are used in chronic infections and often over longer time periods. Unfortunately, in addition to being used in clinical medicine, azoles are employed extensively in agriculture; to control fungal disease in crop plants and intensively-farmed animals. Consequently, azole-resistant Aspergillus can be easily isolated from the environment (Howard & Arendrup, 2011). Aspergillus species are ubiquitous fungal saprotrophs worldwide, and Aspergillus fumigatus is largely responsible for the increased mortality rates of invasive aspergillosis in some immunocompromised patients. Mounting reports of azole resistance in Aspergillus are threatening the effectiveness of clinical and agricultural azole antifungals (Garcia-Rubio et al., 2017). Significantly, the transcription factor that regulates ergosterol biosynthesis in fungi has recently been found to be unique, making it a new potential target for the development of novel antifungals (Yang et al., 2015; Sant et al., 2016; Gumber et al., 2017).
F901318 (olorofim) is a novel antifungal drug that is highly active against Aspergillus species. Belonging to a new class of antifungals called the orotomides that target dihydro-orotate dehydrogenase in the pyrimidine biosynthetic pathway; they stop pyrimidine biosynthesis in fungal cells and thereby block the growth of hyphae. Orotomides are currently in clinical development for the treatment of invasive aspergillosis to overcome this growing problem of resistance to conventional therapies (Oliver et al., 2016; du Pré et al., 2018, 2020). A polycyclic molecule called turbinmicin possesses potent antifungal activity against the multidrug-resistant fungal pathogens Candida auris and Aspergillus fumigatus. The substance is produced by actinobacteria collected from the ocean-dwelling invertebrate sea squirt, Ecteinascidia turbinate. Turbinmicin blocks the action of the protein Sec14p, a protein involved in vesicle trafficking (intracellular transport and localisation of lipid raft proteins) to the hyphal tip (Zhang et al., 2020).
Updated December, 2020