
4.2 Origin of drugs in current use: the diflucan story (contributed by Abrar Malik, 2001)
Abstract
The discovery of Diflucan (fluconazole) was a major landmark in the pharmaceutical industry, as it was the first antifungal drug to be developed that could be used both orally, for minor infections such as candidiasis, or intravenously for more serious systemic infection like cryptococccal meningitis.
Fluconazole development was based on two initial assumptions by the scientists involved: firstly that drugs should be tested in experimental models of infections, and secondly that a polar molecule might have superior pharmacological properties. The developers took these two ideas as a starting point and went through a rigorous process to find the right compound. Eventually they came across 2-(2,4-difluorophenyl)-1,3-bis (1H-1,2,4-triazol-1-yl)-2-propanol, that is fluconazole. This report details the progressive discovery of fluconazole and all the intermediate steps and compounds found along the way. The report also describes the general development of drugs, the history of antifungal agents of the past and examines the major agents currently available. There is also a section devoted to the clinical and pharmacological profile of fluconazole, and any considerations concerning the future development of antifungal agents.
Introduction
The drugs industry plays a major role in modern medicine, as almost every disease and condition known can be prevented, cured or controlled by drugs, and the use of drugs is overwhelmingly preferred to other measures like surgery, which is often used as a last resort due to the potential development of complications.
The increase in the use of drugs has lead to better healthcare, but is problematic. There are many issues raised, especially with the advent of recreational drugs, and whether society is becoming too dependent on drugs to solve our problems.
The range of drugs available is immense, and many more are under development to either improve current medication or provide drugs for use in new areas. This has resulted in a multi-billion pound industry concerned with the discovery, development, production and marketing of drug products. This study focuses on the development of Diflucan (generic name fluconazole), a widely used triazole antifungal agent, and also provides an overview of the history of drugs, current developments and future considerations.
 |
| Fig. 1. Candidiasis of the tongue – one of the several fungal infections for which Diflucan (fluconazole) is an effective treatment [ref. 10]. |
Overview of drug development
It can take from 10-12 years and a cost of $125 million (£88 million) to produce a drug [1] - from the initial discovery right through to its placement on the shelves of a pharmacy. The steps in the process of developing a drug are outlined in Table 1 below.
| 1. Phases of development of pharmaceutical products (from reference 1) | |
| COMPOUND STATUS | STAGE OF DEVELOPMENT |
| Basic research | Discovery of compound, including interaction of chemistry, biochemistry, microbiology, pharmacology and enyzmology |
| Pharmacology | All stages of preclinical investigations, including in vitro and in vivo (animal models) assays, safety assessment in animal models, pharmacology, and formulation studies |
| Clinical Trials: Phase 1 | Clinical pharmacology: first tolerance and pharmacokinetics tests in normal, healthy volunteers |
| Clinical Trials: Phase 2 | Efficacy and safety tests in a limited number of patients with the target disease |
| Clinical Trials: Phase 3 | Controlled clinical trials to determine efficacy and safety in large numbers of patients with the target disease; data are used for registration of the compound with government agency for market approval |
| Market launch | Marketing started in one or more countries |
| Phase 4 studies | Surveillance after approval of drug; based on government request |
| Phase 5 studies | Studies to seek additional marketing claims, new formulations, and combinations |
History of antifungal drug development
In 1665 thrush - caused by Candida albicans - was recorded as a fatal disease [1] and the first microorganism established to cause disease was a fungus in 1835 (Beauvaria bassiana), but despite the fact that fungi were recognized before bacteria, the development of antifungal drugs has been slow. Nystatin, the first major antifungal drug, was not discovered until 1949, and the azole antifungal agents, including Diflucan not until 1969. There may be several reasons for this: fungal diseases are difficult to document due to their nature, and the disease-causing organisms are eukaryotic species which are biochemically similar to human hosts, thus developing an effective drug against the fungus but safe towards the host is difficult. However, there has recently been an increase in the number of immunosuppressed cancer, HIV and organ transplant patients and consequently an increase in the number of opportunistic infections by fungi. Detailed below is a brief history of antifungal drugs [2].
At around the time of the Second World War the treatments available for fungal infections were weak acids and phenolic dyes. Later on in the 1940s a major breakthrough was found in the form of undecylenic acid. Griseofulvin, the first orally effective antibiotic was used for dermatophytosis management in the early 1960s. At around the same time topical agents like tolnaftate and various polyene antibiotics also became available. The late 1960s saw the arrival of broad-spectrum antifungal agents, with the first being iodinated trichlorophenol haloprogin, which functioned by disrupting the fungal cell membrane.
Just before 1970 the broad-spectrum imidazole agents appeared. These act by binding to cytochrome p-450, thus blocking ergosterol synthesis in the fungal cell membrane. Clotrimazole and miconazole were the first imidazoles available and remained the major antifungal agents for years, but recently other compound such as sulconazole, bifonazole, oxiconazole and isoconazole have been introduced. The imidazoles are however ineffective systemically, apart from ketoconazole, but its use has been limited due to its hepatotoxicity and drug interaction effects.
Then came the development of ciclopirox olamine, a pyridone-ethanolamine salt that acts by inhibiting the synthesis of cell membrane proteins. It has been proven to be more effective than clotrimazole. The 1970s saw the development of the triazole antifungal agents that are similar to the imidazoles, but are less likely to cause hepatotoxicity, possibly due to their lesser effects on cytochrome p-450-dependent enzymes. Terconazole was the first triazole developed, followed by itraconazole and fluconazole, the focus of this report.
Newer developments include amorolfine, a dimethylmorpholine agent, used primarily as an agricultural fungicide but may have uses in human fungal infection treatment. Another development is of the allylamine derivatives that act by inhibiting squalene epoxidase and thus block ergosterol synthesis without affecting cytochrome p-450. They have a broad-spectrum of activity, and examples include naftifine and terbinafine.
Development of fluconazole
Before the development of fluconazole the two major antifungal agents available were amphotericin B and flucytosine. The problems with these agents was that amphotericin B has to be given intravenously and had severe adverse effects, whereas flucytosine was orally active but had a limited range of action and resistance was easily developed.
This led to Pfizer Central Research in Sandwich, Kent (UK) commissioning a programme in 1978 to find an agent able to treat life-threatening systemic fungal infections [3], and the subsequent development of fluconazole, 2-(2,4-difluorophenyl)-1,3-bis (1H-1,2,4-triazol-1-yl)-2-propanol.
The task was to find a drug that was safe, have a wide range of action, and be effective both orally and intravenously. This would allow the drug to be used against common infections, such as vaginal candidiasis, dermatomycoses and at the same time also be administered intravenously for seriously ill patients suffering from systemic mycoses.
Experimentation started with azole antifungals because of their mechanism of action of inhibiting ergosterol synthesis, but the disadvantage of azoles was that they are metabolized and are highly lipophilic; this led to their poor oral absorption and low transient blood levels (due to the lipophilic drug binding to proteins). Overcoming these problems would result in an orally effective agent.
Janssen Pharmaceutica announced in mid-1978 that they had prepared ketoconazole, an imidazole derivative, which was proved to be orally effective but was still degraded metabolically [3]. Therefore, Pfizer synthesized a variety of compounds similar structurally to ketoconazole; they included tetrahydrofurans, dithiolanes, dioxolanes and tertiary alcohol derivatives. These compounds showed good potency in vitro but fared less well in animal models of fungal infections.
Efforts were then concentrated on the tertiary alcohol derivatives of imidazole because they displayed better activity in the animal models of infection. Derivatives were then prepared but none displayed better in vivo activity than that of ketoconazole, and were also still susceptible to metabolism.
It was then decided to replace the imidazole unit in the compound with several derivatives, of which the only successful one was the 1,2,4-triazole derivative - it was more active in vivo but four times less potent in vitro.
Consequently, attention was turned to the triazole tertiary alcohol derivatives (Fig. 2), in which the R substituent was varied, in order to obtain compounds resistant to metabolism and of low lipophilicity. The first group used was a 1,2,4-triazole, which resulted in the formation of the bistriazole UK-47,265. This compound performed outstandingly in a mouse model of candidiasis, being almost 100 times more potent than ketoconazole [3].
 |
| Fig. 2. Structure of triazole tertiary alcohol derivatives. |
This led to several bis-triazoles being formed by replacing the dicholorophenyl unit with a variety of chemical substuent groups. This was done via two approaches: firstly the chloroacetyl derivative 1 was reacted with 1,2,4-triazol to yield ketone 2; which was then converted to epoxide 3, and opening of this resulted in the bis-triazole derivative. The second approach was to convert dichloroacetone to dichloropropanol derivative 4, and reacting this with 1,2,4-triazole yielded the bis-triazole. These routes of synthesis resulted in a large number of 2-substituted 1,3-bis-triazolylpropan-2-ol derivatives. These compounds were then tested on murine models of acute systemic candidiasis.
The most potent of these derivatives was 2-phenyl moiety substituted by one or more halogen groups, with the best activity achieved when the halogen group was in position 2 and/or 4. Testing of these compounds then went on to animal models of vaginal candidiasis and dermatomycosis. The four most potent derivatives were then tested pharmacokinetically and for solubility.
It was found that only the 2,4-difluorophenyl analogue was water soluble [3], thus allowing formulation for intravenous administration. This derivative (which was later to be named fluconazole) was then tested in a wide variety of fungal infection models in both normal and suppressed immune system animals. Its activity was exceptional and then its study progressed through safety tests and evaluations in humans before finally being produced and marketed commercially, selling under the trade name of Diflucan in the United Kingdom and United States.
Fluconazole
Fluconazole (Diflucan) is a broad-spectrum antifungal agent available as either tablets for oral administration, a powder for use as an oral suspension; and as a sterile solution for intravenous use. It is known as 2-(2,4-difluorophenyl)-1,3-bis(1H-1,2,4-triazol-1-yl)-2-propanol (Fig. 3), with molecular weight of 306.3 and empirical formula C13H12F2N6O. Its appearance is of a white crystalline solid that is slightly soluble in water and saline [4].
 |
| Fig. 3. Fluconazole [ref. 4]. |
Diflucan tablets contain 50, 100, 150, or 200 mg of fluconazole and the following inactive ingredients: microcrystalline cellulose, dibasic calcium phosphate anhydrous, povidone, croscarmellose sodium, FD&C Red No. 40 aluminium lake dye, and magnesium.
Diflucan for oral suspension contains 350 mg or 1400 mg of fluconazole and the following inactive ingredients: sucrose, sodium citrate dihydrate, citric acid anhydrous, sodium benzoate, titanium dioxide, colloidal silicon dioxide, xanthan gum, and natural orange flavour. After reconstitution with 24 ml of distilled water or Purified Water (USP), each ml of reconstituted suspension contains 10 mg or 40 mg of fluconazole.
Diflucan injection is an iso-osmotic, sterile, nonpyrogenic solution of fluconazole in a sodium chloride or dextrose diluent. Each ml contains 2 mg of fluconazole and 9 mg of sodium chloride or 56 mg of dextrose, hydrous. The pH ranges from 4.0 to 8.0 in the sodium chloride diluent and from 3.5 to 6.5 in the dextrose diluent. Injection volumes of 100 ml and 200 ml are packaged in glass and in Viaflex® Plus plastic containers [4].
Mechanism of action
Fluconazole is fungistatic in action and works by inhibiting fungal cytochrome P-450 sterol C-14 alpha-demethylation, thus obstructing the conversion of lanosterol to ergosterol [5]; which leads to the inhibition of membrane sterol synthesis and therefore preventing fungal cell replication. Another effect is that there is a build up of 14 alpha-methyl sterols and this may increase fluconazole fungistatic effects. It is selectively toxic because in human cells the major sterol is cholesterol rather than ergosterol, and fluconazole (and other triazoles) have lower incidence of side effects because they are much more specific inhibitors of lanosterol alpha-demethylase [6].
Pharmacokinetics
Fluconazole displays pharmacokinetic properties that are similar following administration by the intravenous or oral routes [4]. In normal volunteers, the bioavailability of oral fluconazole is over 90% compared with intravenous. In fasted volunteers, a single oral 400 mg dose leads to a mean peak plasma concentrations (Cmax) of 6.72 mg/ml (range: 4.12 to 8.08 mg/ml) and after single oral doses of 50-400 mg, plasma concentrations and AUC (area under the plasma concentration-time curve) are dose proportional. Peak plasma concentrations (Cmax) in fasted normal volunteers occur between 1 and 2 hours with a plasma elimination half-life of approximately 30 hours (range: 20-50 hours). Steady-state concentrations are reached within 5-10 days following oral doses of 50-400 mg given once daily. But a loading dose on the first day of twice the daily dose results in plasma concentrations close to steady-state by the second day. The volume of distribution (50 to 60 litres) is equal to total body water [5]. Plasma protein binding is low (11-12%). Following either single- or multiple-oral doses for up to 14 days, it penetrates into all body fluids studied (Table 2).
| 2. Fluconazole penetration of tissue/fluid [from reference 4] | |
| Tissue or fluid | Ratio of fluconazole tissue (fluid)/plasma concentration* |
| Cerebrospinal fluid† | 0.5-0.9 |
| Saliva | 1 |
| Sputum | 1 |
| Blister fluid | 1 |
| Urine | 10 |
| Normal skin | 10 |
| Nails | 1 |
| Blister skin | 2 |
| Vaginal tissue | 1 |
| Vaginal fluid | 0.4-0.7 |
| Relative to concurrent concentrations in plasma in subjects with normal renal function. † Independent of degree of meningeal inflammation. | |
In normal volunteers, fluconazole is cleared primarily by renal excretion [4], with approximately 80% of the administered dose appearing in urine as unchanged drug. About 11% of the dose is excreted in the urine as metabolites. The pharmacokinetics are affected by reduction in renal function. There is an inverse relationship between the elimination half-life and creatinine clearance. The dose may need to be reduced in patients with impaired renal function.
Fluconazole's unique pharmacokinetic profile of having a low molecular weight, low plasma binding affinity, water solubility, little first pass metabolism and long half life is what makes it the drug of choice for treating fungal infections.
In vitro activity
Fluconazole in vitro activity is poor. Rogers & Galgiani [cited in reference 1] have reported that at a physiologic pH fluconazole was 16-fold less active than ketoconazole and amphotericin B against yeasts and filamentous fungi when tested in both liquid and solid medium assays. It was also shown to be active in vitro against Candida species and Cryptococcus neoformans, less active against dermatophytes, and totally inactive against Aspergillus species.
Odds et al. [cited in reference 1] examined fluconazole in vitro activity using four different test systems - relative inhibition factors, agar dilution susceptibility assay, effect of drug on hyphal formation of C. albicans and effect on ATP content of C. albicans spheroplasts. Fluconazole was poorly active in all of the assays. Fig. 4 shows that high doses of fluconazole are needed in comparison to other agents to combat in vitro infection.
 |
| Fig. 4. In vitro activity of fluconazole compared with other antifungal agents [from reference 5]. |
In vivo activity
Fluconazole's poor in vitro activity is in contrast to its in vivo activity [1]. Oral administration shows superior activity to ketoconazole and comparable to amphotericin B in treatment of a variety of systemic and superficial fungal infections, including fungal meningitis and Cryptococcus neoformans [1]. Fluconazole was shown to be 20-fold more active in vivo against experimental candidiasis than ketoconazole and is active against tinea versicolor and dermatomycoses. Oral fluconazole has been shown to be active in an animal model of vaginal candidiasis [4].
There are, however, some contradictory data on its activity against experimental aspergillosis [1]. Troke et al. reported it to be 5-20-fold more active than ketoconazole, whereas Graybill showed that it did not protect the mice in the study from aspergillosis. Fig. 5 compares fluconazole to clotrimazole in various infections.
 |
| Fig. 5. Fluconazole versus clotrimazole in fungal infections, showing % of patients who develop fungal infections after prophylaxis with oral fluconazole 200 mg daily, or clotrimazole 10 mg five times a day. Numbers at top of graphs indicate relative hazard (risk of infection or death) between treatments. Graph from reference 5. |
Clinical uses
Fluconazole is effective against:
- Oesophageal and oropharyngeal candidiasis;
- Vaginal candidiasis due to Candida yeast infections;
- Cryptococcal meningitis, especially in AIDS patients;
- Urinary tract infections arising from Candida infections;
- Peritonitis;
- Systemic Candida infections including candiedmia, disseminated candidiasis and pneumonia;
- Prophylaxis (Fig. 6) especially for patients undergoing bone marrow transplantation who receive cytotoxic chemotherapy and/or radiation therapy.
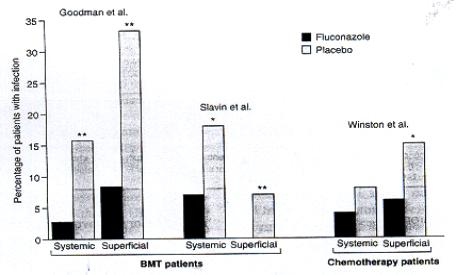 |
| Fig. 6. Fluconazole as prophylaxis, showing % of patients undergoing bone marrow transplantation (BMT) or chemotherapy for acute leukaemia who developed fungal infections during prophylaxis with fluconazole 400mg daily or placebo [from reference 5]. |
Its effectiveness against urinary tract infections is probably due to the high urinary excretion rate of fluconazoles (80% appears as unchanged drug). It is also used as maintenance therapy in AIDs patients with cyptococcal meningitis. There is a continuing debate on the cost-effectiveness of using fluconazole in HIV/AIDs patients in countries with high rates of these infections e.g. Africa. Fluconazole has shown to be ineffective against Candida krusei and Aspergillus, and secondary resistance of Candida spp. seems to be developing [5].
Drug-drug interactions
Fluconazole was tested against several mainstream drugs to test for any interactions and resulting adverse effects:
- Oral contraceptives – there was no significant change in levels of sex hormones after administration of 50 mg daily for 10 days. However higher doses (around 200 mg) have significantly increased levels of circulating levonorgestrel and ethinyl estradiol compared to a placebo [4].
- Cimetidine – Administering 100 mg fluconazole after 400 mg cimetidine resulted in a significant decrease in fluconazole AUC and Cmax, but giving 600 mg cimetidine after 900 mg intravenous fluconazole did not affects it bioavailability or pharmacokinetics [4].
- Antacid – fluconazole has no interactions with antacid administration.
- Hydrochlorothiazide – oral administration of 100 mg fluconazole and 50 mg hydrochlorothiazide resulted in a large increase in fluconazole AUC and Cmax.
- Rifampin – 200 mg of fluconazole administered after 600 mg of rifampin resulted in a decrease in AUC, decrease in half-life and an increase in oral clearance [5].
- Warfarin – 15 mg of warfarin administered after 200 mg of oral fluconazole resulted in a decrease in prothrombin time response.
- Phenytoin – 200 mg of phenytoin followed by 200 mg of fluconazole resulted in a significant increase in phenytoin AUC.
- Cyclosporine – fluconazole caused a significant increase in cyclosporine AUC, Cmax, Cmin and a reduction in oral clearance.
- Zidovudine – fluconazole increases zidovudine AUC after administration of a 200 mg dose.
- Oral hypoglycaemic – fluconazole increased significantly the AUC and Cmax of tolbutamide, glipizide and glyburide [5].
Contraindications and precautions
- Fluconazole should not be administered to patients showing hypersensitivity, but there is no evidence of cross-hypersensitivity with other azole antifungal agents. Clinical trials have shown that fluconazole does not cause carcinogenesis, mutagenesis or fertility impairment [4].
- Care should be taken when administrating fluconazole with the drugs that it has shown to interact with.
Adverse reactions/toxicity
- Patients using fluconazole for single dose therapy can suffer from headaches, nausea and abdominal pain, but they are mild to moderate in severity. Rarely, users may experience diarrhoea, dyspepsia, dizziness, and taste perversion [5].
- Multiple dose therapy patients may also suffer very rarely, from hepatobiliary complaints (ranging from mild transient hepatitis to hepatic failure). Other noted adverse effects are anaphylaxis, seizures and exfoliative skin disorders.
- There has been one reported case of over-dosage with fluconazole, with the patient hallucinating and displaying paranoid behaviour, but his condition was resolved in hospital after 48 hours [4].
Dosage
- Patients using fluconazole for single dose therapy can suffer from headaches, nausea and abdominal pain, but they are mild to moderate in severity. Rarely, users may experience diarrhoea, dyspepsia, dizziness, and taste perversion [5].
- Multiple dose therapy patients may also suffer very rarely, from hepatobiliary complaints (ranging from mild transient hepatitis to hepatic failure). Other noted adverse effects are anaphylaxis, seizures and exfoliative skin disorders.
- There has been one reported case of over-dosage with fluconazole, with the patient hallucinating and displaying paranoid behaviour, but his condition was resolved in hospital after 48 hours [4].
Dosage
Single dose therapy, for example for vaginal candidiasis – 150 mg, as shown in Table 3 below a single 150 mg dose of fluconazole is as effective as multiple doses of other antifungal agents.
| 3. Comparison of clinical effectiveness of antifungal agents in different regimes against vaginal candidiasis, from reference [8]. | |||
| Regimen | Women tested | % Clinical Effectiveness | |
| - 15th day | - 60th day | ||
| Daily 200mg oral itraconazole for 3 days | 50 | 84 | 78 |
| Single 150mg oral fluconazole | 50 | 80 | 76 |
| Daily 100mg intravaginal clotrimazole for 6 days | 50 | 72 | 58 |
Multiple dose therapy for systemic infections - dosage depends on the infecting organism and patient’s response to therapy, but a typical regimen is a 200 mg dose on the first day followed by 100 mg once daily [4].
of antifungal agents
Table 4 shows the currently available major antifungal drugs for clinical use. Their chemical structure, mechanism of action, treatment indications and toxic effects are also shown for comparison:
| 4. Major antifungal drugs [From reference 9] | ||||
| Antifungal agent | Chemical structure | Probable mechanism (s) of action | Indications for treatment of deep mycoses and/or etiologic fungi | Principal toxic effects |
| Amphotericin B | Polyene macrolide | Combination with sterols, especially ergosterol in the cytoplasmic membrane, to establish pores in cell membrane; also oxidative membrane damage | Aspergillus spp., Candida spp., Cryptococcus neoformans, Histoplasma formans, Histoplasma capsulatum, Blastomyces dermatitidis, Coccidioides immitis, Trichosporon beigelii, Fusarium spp., Bipolaris, Exersohilum | Nephrotoxicity: azotemia, renal tubular acidosis hypokalemia, hypomagnesemia, bone marrow suppression; fever and shaking chills during infusion thrombophlebitis |
| Flucytosine | fluorinated pyrimidine | 5-fluorocytosine, 5-fluorouridine and their phosphorylated nucleotides cause inhibition of fungal thymidylate synthetase and DNA synthesis; 5-fluorouridine triphosphate inhibits RNA processing | Use in combination with amphotericin B for deep mycoses due to Cryptococcus neoformans, some dematiaceous hypho- mycetes, and Candida spp. | Bone marrow suppression diarrhea hepatotoxicity cerebellar defects
|
| Miconazole | N-substituted imidazole | inhibition of synthesis of membrane sterols at the level of oxidative 14-C demethylation through inhibition of yeast cytochrome P-450 dependent enzymes | Pseudallescheria boydii | pruritis, headache, thrombophlebitis headache thrombophlebitis hepatotoxicity |
| Ketoconazole | N-substituted imidazole | similar to that of miconazole | non-meningeal infections due to Paracoccidioides braziliensis, chronic cavitary histoplasmosis, disseminated histoplasmosis in clinically stable non immunosuppressed patients, non-meningeal blastomycosis and certain forms of coccidioidomycosis (cutaneous lesions, draining sinus tracts, soft tissue abscesses, non-cavitary pulmonary infiltrates, synovitis and osteomyelitis) | nausea, vomiting, anorexia, hepatoxicity: usually transient; rarely icteric endocrinologic; gynecomastia; diminished libido; rarely, adrenal insufficiency |
| Itraconazole | N-substituted triazole | similar to that of miconazole | indications similar to those for ketoconazole with possible addition of sporotrichosis and selected conditions of aspergillosis | less gastrointestinal toxicity than ketoconazole; possible Aldosterone-like effect |
| Fluconazole | N-substituted bis-triazole | similar to that of miconazole | Cryptococcal meningoencephalitis | infrequent nausea; infrequent hepatotoxicity (asymptomatic transaminase elevation) |
Conclusion
The initial development of Diflucan (fluconazole) was done with two assumptions in mind: that compounds should be tested for activity in experimental infections, not just for susceptibility of isolates in vitro; and that a polar molecule might have superior pharmacological properties [7]. This lead to an extremely long and rigorous process of hypothesizing, formulating and testing compound after compound to find an antifungal drug that would function both orally and intravenously, and thus be used against minor topical infections and more severe systemic infections.
Eventually the right compound was found and marketed as Diflucan (fluconazole). It displayed pharmacological properties like no other antifungal drug before it and accomplished what they set out to achieve. Its development would have cost millions of pounds and involved many hours and scientists, yet the end result would be historical within the pharmaceutical industry.
Fluconazole is not perfect; there are several fungi on which it has no effect, and there seems to be resistance developing in certain Candida species. Current literature suggests that antifungal drug production has been generally slow moving and that there is room for improvement, and that any future research and development into this field would have to take in the following considerations:
The drug needs to display a potent, broad spectrum of activity against fungal pathogens.
- flexibility of parental or oral administration it required;
- should be non-toxic;
- must display favourable pharmacokinetics, including easily measured serum and urine levels, effective penetration into all tissue compartments, complete bioavailability and predictable adjustment, of dosage in renal or hepatic insufficiency;
- must take into account the role of host defence mechanisms against opportunistic fungi, such as interferons, interleukins and colony stimulating factors.
There is hope that one day a drug will be developed that satisfies all the above criteria, and that it can eclipse the success of Diflucan (fluconazole). It has not been found yet.
References
1. Fromtling, R.A. (1988). Overview of medically important antifungal azole derivatives. Clinical Microbiology Reviews, 1: 187-217.
2. Smith, E.B. (1990). History of antifungals. Journal of the American Academy of Dermatology, 23: 776-778.
3. Richardson, K., Cooper, K., Marriott, M.S., Tarbit, M.H., Troke, P.F. & Whittle, P.J. (1990). Discovery of fluconazole, a novel anitfungal agent. Reviews of Infectious Diseases, 12 Suppl 3: 267-271.
4. Pfizer US Webpage http://www.pfizer.com/hml/pi’s/diflucanpi.html
5. Goa, K.L. & Barradell, L.B. (1995). Fluconazole, an update of its pharmacodynamic and pharmacokinetic properties and therapeutic use in major superficial and systemic mycoses in immunocompromised patients. Drugs, 50: 658-90.6. Neal, M.J. (1997). Medical Pharmacology at a Glance. 3rd edition. Blackwell Science.
7. Bodey, G.P. (1992). Azole anifungal agents. Clinical Infectious Diseases, 14 Suppl 1: 161-169.
8. Mikamo, H., Kawazoe, K., Sato, Y., Hayasaki, Y. & Tamaya, T. (1998). Comparative study on the effectiveness of antifungal agents in different regimens against vaginal candidiasis. Chemotherapy, 44: 364-368.
9. Walsh, T.J. & Pizzo, A. (1988). Treatment of systemic fungal infections: recent progress and current problems. European Journal of Clinical Microbiology and Infectious Diseases, 7: 460-475.
10. Underwood, J.C.E. (1996). General and Systematic Pathology. 2nd edition. Churchill Livingstone.
Updated December 7, 2016