
4.4 Origin of drugs in current use: the story of statins (contributed by Mark Gilson, 2000)
Abstract
The story of the statin based drugs provides an interesting insight into the discovery and development of modern pharmaceuticals. As well as looking at the complexity of the technology and science involved, this Special Studies Module also looks at the time and resources that must be devoted to developing a marketable drug from an initial concept. Whilst the pharmacology of a specific family of drugs might be similar, small differences in molecular composition, and human physiology can substantially alter the overall effectiveness of the drug, and even classes of patient in which the drug might be of use.
Given the cost of development, many drugs will become unaffordable to a large proportion of the world population, even if the cost seems reasonable in terms of suffering and the palliative care that might have to be offered in their place. The problem of cost may also be exasperated as the development and marketing of new drugs often seems to be in the control of a handful of large multinational companies.
Discovery of the statins
Various cholesterol-lowering agents had been discovered during the 1950s and 1960s. However, the majority had unwanted side effects. In 1971 Dr Akira Endo and Dr Masao Kuroda began the search for a cleaner drug to treat hypercholesterolemia [1]. The starting point for their research was work that had previously been carried out during the 1960s.
Various experiments on animals and humans had shown that cholesterol could either be absorbed from the diet, or if the diet was lacking sufficient cholesterol to meet the body's needs, then it could be synthesized; mainly in the liver (82%) and the intestine (11%) [2]. However, if the diet was rich in cholesterol then synthesis within the body virtually stopped. Previous work had shown that cholesterol production within the body was controlled by a feedback mechanism in which cholesterol inhibited the enzyme β-hydroxy-β-methylglutaryl-CoA reductase (HMG Co-A reductase). By inhibiting this enzyme, the conversion of HMG-CoA to mevalonic acid was stopped [3] - this step being crucial to the body in creating cholesterol.
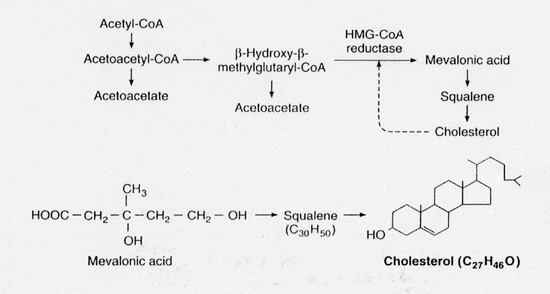 |
| Fig.1. Pathway of cholesterol synthesis [from reference 4]. |
The aim of Drs Kuroda and Endo was therefore to find a substance that would inhibit the action of HMG-CoA reductase. In looking to the microbial world, researchers were hoping to find a microorganism that produced an HMG-CoA reductase inhibitor as a defence mechanism against attack by other microbes which relied on sterols as part of their biochemical make up.
The search for a suitable compound took two years and involved more than 6,000 microbes. Initially, researchers were looking for microbial broths that would inhibit the incorporation of carbon-14 acetate into lipids. In a second set of experiments, broths that had acted as inhibitors between acetate and lipid were tested to see whether they would inhibit the production of lipids from tritium-labelled mevalonate. Broths that acted as inhibitors in the first set of experiments, but not in the second set of experiments, were deemed to be inhibitors in the early part of cholesterol synthesis.
In the early stages, two moulds were found to meet the above requirements. Firstly Pythium ultimum was found to produce a substance called citrinin. Citrinin, also an anti-fungal, was shown to irreversibly inhibit HMG Co-A reductase. The second mould shown to inhibit lipid synthesis was Penicillium citrinum. The active compound from P. citrinum was isolated by silica gel chromatography and crystallization. Further investigation of the inhibiting compound by spectroscopy, X-ray crystallography and chemical means showed the new compound, ML-236B (now known as mevastatin) to have the following structure:
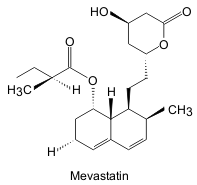 |
| Fig. 2. Structure of mevastatin [redrawn after reference 1] |
Research into mevastatin showed that the compound was capable of inhibiting lipid synthesis from either carbon-14 HMG Co-A, or carbon-14 acetate. However, there was no inhibitory effect on lipid production from tritium-labeled mevalonate. From this, it was possible to deduce that mevastatin did in fact inhibit the enzyme HMG Co-A reductase.
By 1976 the drug company Merck & Co. had become involved in the development of the statins. Having successfully repeated the experiments of Endo and Kuroda, they proceeded to isolate a similar molecule, lovastatin, from Aspergillus terreus. The new compound was slightly more effective as an HMG Co-A reductase inhibitor than mevastatin. The time periods involved in getting new drugs to the market are also well demonstrated by lovastatin. Although Merck isolated the new compound in 1978, it did not receive approval from the US Food and Drug Administration until 1987.
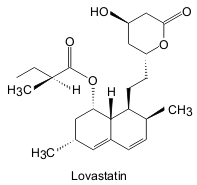 |
| Fig. 3. Structure of lovastatin [redrawn after reference 1] |
Development of other drugs based on mevastatin and lovastatin has continued all over the world. Three main approaches have been utilized. Firstly, synthetic compounds, such as fluvastatin, were produced. Research in this area concentrated on replacing the decalin ring of the fungal compounds with an aromatic ring. Secondly, chemical alteration of fungal products created drugs such as simvastatin. In this drug, modifications were made to the acyl group at C1. Finally, microbial alteration of fungal compounds has lead to drugs such as pravastatin.
By altering the basic chemical composition of the mevastatin molecule, potency of the drug can be increased. Simvastatin is approximately twice as potent as pravastatin and lovastatin, whilst mevastatin is the least powerful. However, in changing the shape of the active molecule, the chances and severity of side effects is also altered. For example there is an increased risk of muscle toxicity with lovastatin, in comparison to pravastatin.
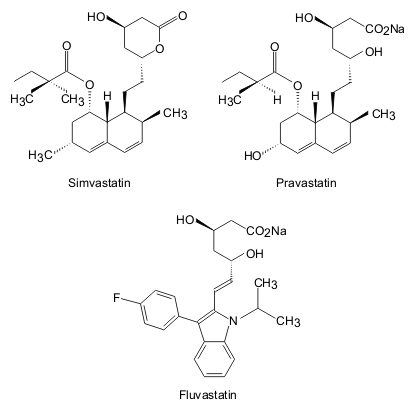 |
| Fig. 4. Chemical structures of three statins, including the synthetic structural analogue, Fluvastatin [redrawn after reference 1]. |
Pharmacology and clinical aspects of the statins
The production of mevalonic acid (a precursor to cholesterol) is brought about when HMG Co-A binds to the enzyme HMG Co-A reductase. After this has occurred, NADPH binds to the enzyme/substrate combination. A reaction then occurs in which NADPH is oxidized to NADP-CoA, and HMG Co-A is reduced to mevalonic acid. As the affinity of HMG Co-A reductase is substantially higher for the statins (in the case of mevastatin 10,000 times higher) than it is for HMG Co-A, mevastatin acts as a reversible competitive inhibitor to the enzyme reaction and less mevalonic acid is produced in its presence. Thus the cholesterol production pathway is broken.
The introduction of a competitive inhibitor for HMG Co-A reductase results in two physiological responses. In compensation for the inhibition, cells begin to produce more HMG Co-A. The direct reduction in circulating cholesterol is therefore only small. However, the number of low-density lipoprotein (LDL) receptors on hepatocytes increases markedly[5]. As the liver is responsible for removing LDLs (of which cholesterol is a component) from plasma via the LDL receptor mechanism, blood cholesterol levels also fall dramatically.
As LDL receptors are critical to the effectiveness of the statins in reducing plasma cholesterol, it is worth noting their ineffectiveness in treating patients with homozygous familial hypercholesterolaemia. In this disease, the patient will have inherited recessive and ineffective alleles for LDL receptor production from both parents. The lack of LDL receptors means that cholesterol clearance from blood plasma cannot be increased by this method. Therefore the statins are of little use. One exception to this rule is Atorvastatin. Atorvastatin has been shown to be of use in patients with homozygous familial hypercholesterolaemia as it has a much longer inhibitory effect on the HMG Co-A reductase enzymes with which it comes into contact [6]. Refinement of the mevastatin molecule has therefore increased the range of patients for whom the statins are useful.
Although some early research carried out in rodents indicated that the statins would only work in the very short term, more positive trials in dogs, monkeys and man have shown them to be potent cholesterol lowering drugs. One such trial was the Scandinavian Simvastatin Survival Study [6]. In the study 4444 patients who had recovered from heart attacks, or who suffered from angina and had plasma cholesterol levels of between 5.5 and 8 mmol/litre were split into two groups. One of the groups was given Simvastatin, and the other group a placebo. Treatment was continued for a median 5.4 years. In the group treated with Simvastatin, cholesterol levels were lowered by 35%, and death from coronary disease was reduced by 42%.
Side effects resulting from treatment with the statins are uncommon, although concerns were raised as a result of trials carried out in Japan during the 1980s. In this trial toxic effects were reported to have occurred in dogs [1]. However, the research was largely discredited as the dogs had been given a dose of mevastatin 1000 times greater than that shown to be effective in man. Further research involving pravastatin and a placebo showed that only 1.7% of patients receiving pravastatin were withdrawn from therapy during a four month trial, in comparison to 1.2% of patients who were receiving the placebo [7]. Known side effects of the statins include gastrointestinal upset, insomnia and rashes. However, more serious side effects may include myositis, hepatitis and renal failure [6].
Whilst the statins are of use in treating hypercholesterolaemia caused by LDLs, they are of less use in treating other hyperlipoproteinaemias. The effect of statins on very low-density lipoproteins (VLDL) and plasma triglycerides is limited, and they may even cause a slight increase in plasma concentrations of high-density lipoproteins (HDL). However, given that high plasma concentrations of LDL-cholesterol is a greater risk factor in the development of atherosclerosis, and therefore coronary heart disease (CHD), their importance in the fight to reduce deaths should not be underestimated[8].